Researchers Design and Build First Artificial Protein
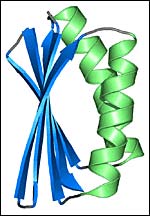
A computer-generated image of the artificial protein, Top7 <br>Image: Gautam Dantas/University of Washington
Using sophisticated computer algorithms running on standard desktop computers, researchers have designed and constructed a novel functional protein that is not found in nature. The achievement should enable researchers to explore larger questions about how proteins evolved and why nature “chose” certain protein folds over others.
The ability to specify and design artificial proteins also opens the way for researchers to engineer artificial protein enzymes for use as medicines or industrial catalysts, said the study’s lead author, Howard Hughes Medical Institute investigator David Baker at the University of Washington.
Baker and colleagues Brian Kuhlman, who is now at the University of North Carolina, Chapel Hill, and graduate student Gautam Dantas at the University of Washington, published their studies in the November 21, 2003, issue of the journal Science. The scientists collaborated on the studies with other researchers at the University of Washington and the Fred Hutchinson Cancer Research Center in Seattle.
Proteins are initially synthesized as long chains of amino acids and they cannot function properly until they fold into intricate globular structures. Understanding and predicting the rules that govern this complex folding process — involving the folding of the main backbone and the packing of the molecular side chains of the amino acids — is one of the central problems of biology.
According to Baker, the ability to specify a desired folded protein structure and then to create that protein offers powerful scientific and practical benefits. “First, specifying a protein fold and then designing that protein is a very stringent test of our current understanding of the forces and energetics of macromolecular systems,” he said. “Because designing something that’s completely new means you can’t copy any aspect from nature.
“Secondly, if one can design completely new structures, one can potentially design novel molecular machines — proteins for carrying out new functions as therapeutics, catalysts, etc. And finally, there’s the evolutionary question of whether the folds that are sampled in nature are the limit of what’s possible; or whether there are quite different folds that are also possible,” he said. “Basically, we want to understand whether nature only sampled a subset of what’s possible,” said Baker.
The challenges of designing an amino acid sequence to fold into a new structure were considerable, said Baker. “If you draw on the back of an envelope some arbitrary protein structure, it might be that there is simply no amino acid sequence that will fold up to that structure. We had to develop methods to computationally sample possible structures similar to the one drawn on the back of the envelope, searching for a conformation for which there exists a very low energy amino acid sequence,” he said.
Baker and his colleagues took advantage of methods for sampling alternative protein structures that they have been developing for some time as part of the Rosetta ab initio protein structure prediction methodology. “Indeed, the integration of protein design algorithms (to identify low energy amino acid sequences for a fixed protein structure) with protein structure-prediction algorithms (which identify low energy protein structures for a fixed amino acid sequence) was a key ingredient of our success,” Baker said. He likened the problem to the three-dimensional version of attempting to create a specified outline for a jigsaw puzzle, given only a certain number of pieces — the equivalent of the 20 known amino acids in nature. In addition, he said, these amino acids can rotate into a number of different conformations.
“At each position, you can have one of the twenty amino acids, and for each of those amino acids you can have on the order of ten different shapes,” he said. “So, you have two hundred different possible shapes for each piece. With those restrictions, it may be that there are some outlines to this jigsaw puzzle that you just cannot achieve. So you need to have a way of changing the boundary to find a protein that can actually be made, because the main constraint is that the side chains fit together perfectly in the interior of the protein.
“Thus, the problem is that the number of alternatives can be huge. Even for one fixed backbone conformation, you have an astronomical number of possible amino acid sequences,” said Baker. “So, we needed a computational approach to search the huge space of possible conformations and possible amino acid sequences efficiently.”
In their design and construction effort, the scientists chose a version of a globular protein of a type called an alpha/beta conformation that was not found in nature. “We chose this conformation because there are many of this type that are currently found in nature, but there are glaring examples of possible folds that haven’t been seen yet,” he said. “We chose a fold that has not been observed in nature.”
Their computational design approach was iterative, in that they specified a starting backbone conformation and identified the lowest energy amino acid sequence for this conformation using the RosettaDesign program they had developed previously RosettaDesign is available free to academic groups at www.unc.edu/kuhlmanpg/rosettadesign.htm.
They then kept the amino acid sequence fixed and used the Rosetta structure prediction methodology they had previously used successfully for ab initio protein structure prediction to identify the lowest energy backbone conformation for this sequence. Finally, they fed the results back into the design process to generate a new sequence predicted to fold to the new backbone conformation. After repeating the sequence optimization and structure prediction steps 10 times, they arrived at a protein sequence and structure predicted to have lower energy than naturally occurring proteins in the same size range.
The result was a 93-amino acid protein structure they called Top7. “It’s called Top7, because there was a previous generation of proteins that seemed to fold right and were stable, but they didn’t appear to have the perfect packing seen in native proteins,” said Baker.
The researchers synthesized Top7 to determine its real-life, three-dimensional structure using x-ray crystallography. As the x-rays pass through and bounce off of atoms in the crystal, they leave a diffraction pattern, which can then be analyzed to determine the three-dimensional shape of the protein.
“One of the real surprises came when we actually solved the crystal structure and found it to be marvelously close to what we had been trying to make,” said Baker. “That gave us encouragement that we were on the right track.”
According to Baker, the achievement of designing a specified protein fold has important implications for the future of protein design. “Probably the most important lesson is that we can now design completely new proteins that are very stable and are very close in structure to what we were aiming for,” he said. “And secondly, this design shows that our understanding and description of the energetics of proteins and other macromolecules cannot be too far off; otherwise, we never would have been able to design a completely new molecule with this accuracy.”
The next big challenge, said Baker, is to design and build proteins with specified functions, an effort that is now underway in his laboratory.
Media Contact
More Information:
http://www.hhmi.org/news/baker3.htmlAll latest news from the category: Information Technology
Here you can find a summary of innovations in the fields of information and data processing and up-to-date developments on IT equipment and hardware.
This area covers topics such as IT services, IT architectures, IT management and telecommunications.




Newest articles

NASA: Mystery of life’s handedness deepens
The mystery of why life uses molecules with specific orientations has deepened with a NASA-funded discovery that RNA — a key molecule thought to have potentially held the instructions for…

What are the effects of historic lithium mining on water quality?
Study reveals low levels of common contaminants but high levels of other elements in waters associated with an abandoned lithium mine. Lithium ore and mining waste from a historic lithium…

Quantum-inspired design boosts efficiency of heat-to-electricity conversion
Rice engineers take unconventional route to improving thermophotovoltaic systems. Researchers at Rice University have found a new way to improve a key element of thermophotovoltaic (TPV) systems, which convert heat…